


ROSAİ-DORFMAN HASTALIĞI: VAKA SUNUMU
2Şişli Etfal Eğitim ve Araştırma Hastanesi, Patoloji Labaratuvarı, İstanbul, Türkiye
Özet
Rosai-Dorfman Hastalığı sebebi bilinmeyen nadir bir hastalıktır. Spontan remisyon ile ölümcül vital organ tulumu arasında değişen klinik spektruma sahip olan bu hastalık sıklıkla baş ve boyun bölgesini tutar. Bu yazıda baş boyun bölgesini tutan Rosai-Dorfman hastalığı tanısı almış genç bir hastanın klinik takibi sunulmuş ve literatür eşliğinde tartışılmıştır.Giriş
Rosai-Dorfman Hastalığı (RDH) sebebi bilinmeyen nadir bir hastalıktır. İlk kez 1969 yılında Rosai ve Dorfman tarafından tanımlanmıştır.[1] Spontan remisyon ile ölümcül vital organ tutulumu arasında değişen klinik spektruma sahip olan bu hastalık sıklıkla baş ve boyun bölgesini tutar. [2] Bu yazıda baş boyun bölgesini tutan Rosai-Dorfman hastalığı tanısı almış genç bir hastanın klinik takibi sunulmuş ve literatür eşliğinde tartışılmıştır.Olgu Sunumu
16 yaşında erkek hasta yaklaşık 3 aydır olan ağrısız, sert, boyun kitlesi ile başvurdu. Şikayetine yandaş semptomlar eşlik etmiyordu. Medikal hikayesinde bir özellik yoktu. Boyun muayenesinde sol taraf posterior üçgende en büyüğü 3x2cm olmak üzere multipl semimobil, palpasyonla ağrısız, üzerinde ısı artışı, hiperemisi olmayan fluktuasyon vermeyen lenfadenopatiler mevcuttu. Bunun dışındaki otorinolaringolojik muayenesinde ve fizik muayenesinde bir özellik saptanmadı. Laboratuar tetkiklerinde sedimantasyon; 1 saatte 12mm ve 2 saatte 25mm olup diğer tetkikleri de normal sınırlardaydı. Hastaya yapılan boyun ultrasonografisinde; Sol posterior üçgende en büyüğü 19x8.8mm olmak üzere toplam 4 adet kistik natürde hilusa ait ekojenite izlenen öncelikle benign formda lenfadenopati izlenmiştir ve ayrıca sağ juguler zincirde en büyüğü 15x7mm boyutlarında birkaç adet benign formda lenfadenopatiler izlenmiştir şeklinde rapor edildi. Sol posterior üçgendeki kitleden yapılan ince iğne aspirasyon biyopsi (İİAB) sonucu şüpheli, düşük grade lenfoma ve lenfadenit ayrımı için lenf bezi eksizyonu önerilir şeklindeydi (Şekil 1). Hastaya bunun üzerine genel anestezi altında eksizyonel biyopsi planlandı. Operasyon sırasında kitlenin sol posterior üçgende kapsüllü olduğu ve çevre dokulardan kolay ayrıldığı gözlendi. Tamamı çıkarılan kitle spesmen olarak patolojiye gönderildi ve CD68 ile immünhistokimyasal boyama sonucu parakortikal nodüler histiositosiz, Rosai-Dorfman hastalığı ile uyumludur olarak rapor edildi (Şekil 2). Kitlenin bütün olarak çıkarılmasından ve boyunda başka patolojik lenfadenopati ve kitle olmadığı için hastaya ek bir tedavi modalitesine gerek olmadığı ve klinik takibin uygun olacağına karar verildi. Hasta önce aylık, daha sonra üç aylık kontrollere çağrıldı. Operasyondan sonra 15 ay takip edilen hastada nüks yönünde herhangi bir bulguya rastlanmamıştır.
 | İnce iğine aspirasyon biopsisinde, yaymalarda değişik maturasyonlarda lenfositler, arada seyrek histiositler, zeminde eritrositler görüldü. HE X 200 |
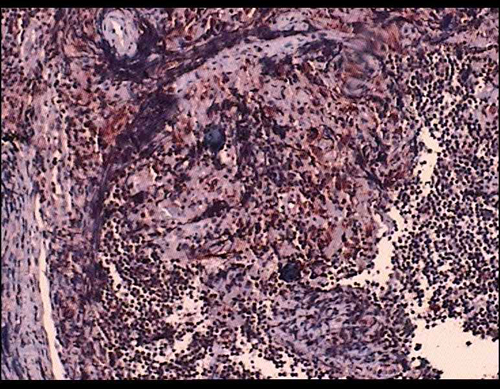 | Lenf bezinde sinüslerde yaygın lenfosit varlığını gösteren, immünohistokimyasal olarak CD68 ile pozitif boyanan hücreler. CD68 X 100 |
Tartışma
Çocukluk çağı histiositozları; monosit makrofaj ve/veya dentritik hücre sisteminde, lokalize veya generalize hücre proliferasyonu ile karakterize bir grup hastalıktır.3 RDH Histiositozis X, Hemofagositik sendrom, Juvenile xanthogranulomatoz ve Malign Histiositoz gibi bir çocukluk çağı histiositozudur. İlk kez 1969 yılında Rosai ve Dorfman tarafından tanımlanmış; aynı otörler 1972 yılında hastalığın diğer özelliklerini tanımladıkları 34 vakalık serilerini sunmuşlardır.[4]Doğu ülkelerinde batıya göre daha sık olarak görülen hastalık, sıklıkla ilk 2 dekadda görülür ve erkek oranı daha fazla gibi görünmektedir[2,5].
Hastalığın etiolojisi bilinmemektedir. İmmünite yetersizliği, otoimmünitenin veya neoplastik prosesin bu hastalığın nedeni olduğuna dair destekleyici bir bulgu yoktur. Ancak hastalık poliartralji, glomerulonefrit ve tip I diabetes mellitus gibi otoimmün patolojilerle birlikte görülebilir.[2] RDH gelişiminde enfeksiyöz orijin öne sürmüş ve bu öngörü RDH tutulumu olan dokularda mikobakteri üretilmesi ile ortaya konmuştur. Ayrıca EBV ve HPV 6 da sorumlu tutulmuş ancak bu öngörü kanıtlanamamıştır.[6]
Hastalık %75-87 oranında baş boyun bölgesinde görülür.[2,6] Tipik olarak boyunda masif, iki taraflı, ağrısız lenf nodu büyümesi olarak karşımıza çıkar. Extranoduler tutulum % 30-43 arasındadır ve yine sıklıkla baş ve boyun bölgesinde görülür.[7] Baş boyun bölgesinde; deri nazal kavite, paranazal sinüs, nazofarenks, tiroid gland, parotis, submandibular gland, larenks, temporal kemik ve nadiren de orbita tutulabilir[2,7,8] ve klinik hastalığın tutulum bölgesine göre değişkenlik gösterir. Kemik tutulumu da görülebilir. Başboyun bölgesi tutulumu olanların % 68inde başka bir lenfoid olmayan sahada da tutulum mevcuttur.[6] Baş boyun dışı tutulabilecek diğer organlar akciğer, böbrek, intestinal sistem, karaciğer ve pankreastır. Yine nadir olarak santral sinir sistemi tutulumu görülebilir[9]. Beraberinde ateş atakları ve kilo kaybı görülebilir. Spesifik laboratuar bulgusu olmamasına rağmen mikrositik anemi, artmış eritrosit sedimantasyon hızı, poliklonal hipergamaglobulinemi [2,6,7] , artmış Ig G düzeyleri [6] trombositoz ve CD4/CD8 düzeylerinde değişme [7] görülebilir. Hastamız erkek ve 2. dekatta olup ayrıca tutulum yeri olarak baş-boyun bölgesi olmasıyla hastalığın genel karakteristik özelliklerini taşımaktadır. Ancak hastada sedimentasyon yüksekliği dışında patolojik laboratuar bulgusu saptanmamıştır.
İİAB tanısal değeri kısıtlıdır ve literatürde RDH tanısının İİAB ile konduğu az sayıda vaka mevcuttur. Tanı için genelde açık biyopsi gerekir.[6] Tanı konması lenf nodu tutulumu olmayan ekstranodüler hastalık durumunda zorlaşır ve bu durumda çoklu biyopsiler gerekebilir.[7,10] Hastamızda da boyunda kitleye ayırıcı tanı amacıyla rutin tahliller yapıldıktan sonra İİAB yapılmış ancak patolojik tanısı tam olarak yapılamamıştır. Kesin tanı ancak eksizyonel biyopsiden sonra konulabilmiştir. Bizim vakamızda İİAB sonucu eksizyonel biyopsi önerilmesi üzerine hastaya eksizyonel biyopsi uygulanmış ve tanı bu şekilde konmuştur.
Lenf nodlarının histopatolojik incelemesinde makroskopik olarak belirgin fibrozis mevcuttur ve kesit yüzeyi, içerdiği yağ miktarı ile bağlantılı olarak, yeşilden sarıya değişir. Mikroskopik olarak lenf sinüslerinde genişleme mevcuttur. Lenfositler, histiositler ve çoğunlukla geniş veziküler nükleuslu yoğun miktarda lipid içeren şeffaf sitoplazmalı histiositlerle doludur. Bu histiositlerin çoğu sitoplazmalarında lenfositler içerir. Bu durum emperipolezis veya Rosai Dorfman hücreleri olarak adlandırılır. Emperipolezis spesifik olmamakla beraber RDH tanısında belirgin bir özelliktir ve önemli bir tanısal değere sahiptir. Ayrıca sinüs histiositlerin önemli bir özelliği de içerdikleri sitoplazmik yağdır. Bu yağ içeriğinin S-100 ile kuvvetli reaktifliği bulunup tanıda yardımcı bir faktördür. [10] Ekstranodal hastalık durumunda fibrozis daha belirgindir ve daha az sayıda Rosai Dorfman hücresi içerir. Ekstranodal hastalık durumunda tanı konması daha da zorlaşır ve tanı için multipl biyopsiler gerekebilir. [6,7]
İmmünohistokimyasal olarak RDH diğer histiositik proliferasyonlardan ayrıldığı söylenebilir. RDH güçlü S-100 pozitifliği mevcuttur[7]. Ayrıca HAM 56, CD14, CD68, ve MAC 387 gibi makrofaj belirteçleri pozitiftir. [2]
RDH ayırıcı tanısında pek çok hastalık mevcuttur. Wegener granulomatoz, malin retiküloz eozinofilik granülom, Hodgkin ve fibroinflamatuar değişiklikler RDH ile karışabilir ancak bu hastalıklarda emperipolezis yoktur. Wegener Miculicz hücrelerinin varlığı ve Warthin Starry gümüş boyamayla RDH ayrılır. Yine Wegener Hastalığında ANCA pozitiftir ve S-100 reaktivitesi bulunmaz.[2] Hodgkin hastalığında lenf nodu tutulumu olmadan ekstranodal alanların tutulumu nadirdir ve Hodgkin hastalığında Reed Stenberg hücreleri pozitiftir. Eozinofilik granülom S-100 pozitifliği ve emperopolezis birlikte görülebilir. Ayırım eozinofilik granülomdaki langerhans hücrelerinin içerdiği Birbeck granüllerinin elektron mikroskop ile gösterilmesi ile konulabilir.[2] Ayrıca Eozinofilik Granülom CD1a pozitif olup, RDH genelde CD1a negatiftir. Metastatik kanserler ve malign melanomdan HM 45 ve pankeratin negatifliği ile ayrılır.[5] Ayrıca lenfoma da RDH ile karışabilir.
Hastalığın tedavisinde, spontan remisyon sık olduğundan, gözlem esastır. Hiçbir tedavi modalitesinin üstünlüğü yoktur. Antibiyotikler, asiklovir, steroidler, radyoterapi ve kemoterapi tedavide kullanılmıştır[5]. Tedavide ayrıca yüksek doz Thalidomide kullanımı da seçenekler arasındadır. [7] Cerrahi kozmetik deformite ve hayatı tehdit eden veya fonksiyonel bozukluğa yol açan durumlarda endikedir. Vinca alkaloidi, alkile edici ajanlar ve kortikosteroid rejiminin etkili olduğu da bildirilmiştir. [3]
Hastalığın prognozu genelde iyi olup; eşlik eden immünolojik anormallik, genç yaş, ekstranodal tutulum (özellikle böbrek, karaciğer) kötü prognostik kriterlerdir. [3]
Biz hastamıza eksizyonel biyopsi uyguladıktan sonra başka bir tedavi modalitesi uygulamayı düşünmedik ve klinik olarak takip ettik. Operasyon sonrası 15 ayda halen nüks yönünde bir bulgu yoktur. Hastamızda daha önceden tarif edilen kötü prognoz kriterleri, genç yaş dışında yoktu.
Kaynaklar
1) Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy: a newly recognized benign clinicopathologic entity. Arch. Pathol. 1969:87;6370. [ Özet ]
2) Cocker RS, Kang J, Kahn LB. Report of a Case Presenting as a Midline Thyroid Mass. Arch Pathol Lab. 2003;127:197-200, [ Özet ]
3) Unal OF, Koybasi S, Kaya S. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease). Int J Pediatr Otorhinolaryngol. 1998:44;173-6, [ Özet ]
4) J. Rosai and F.R. Dorfman, Sinus histiocytosis with massive lymphadenopathy: A pseudolymphomatous benign disorder. Cancer 1972:30;117488. [ Özet ]
5) Lu CI, Kuo TT, Wong WR, Hong HS. Clinical and histopathologic spectrum of cutaneous Rosai-Dorfman disease in Taiwan. J Am Acad Dermatol. 2004 Dec;51(6):931-9, [ Özet ]
6) Ahsan SF, Madgy DN, Poulik J. Otolaryngologic manifestations of RosaiDorfman disease. Int J Pediatr Otorhinolaryngol. 2001:59;221-7, [ Özet ]
7) Unal OF, Kocan EG, Sungur A, Kaya S. Rosai Dorfman disease with multi organ involvement in head and neck region. Int J Pediatr Otorhinolaryngol. 2004:68;581-4, [ Özet ]
8) Gassel AM, Lamskemper DM, Sold-Darseff J, Muller JG, Muller-Hermelink HK. Isolated Rosai-Dorfman disease. Pathologe. 2004 Sep;25(5):398-401, [ Özet ]
9) Ture U, Seker A, Bozkurt SU, Uneri C, Sav A, Pamir MN. Giant intracranial Rosai-Dorfman disease. J Clin Neurosci. 2004 Jun;11(5):563-566, [ Özet ]